Tianjin Medical Journal ›› 2023, Vol. 51 ›› Issue (7): 681-686.doi: 10.11958/20230447
Special Issue: 专题研究
• Monograph·Amyotrophic Lateral Sclerosis • Previous Articles Next Articles
LIU Yufei( ), ZHU Ju, LIU Na, REN Yanping, SUN Xiaohui, TIAN Li(), ZHANG Zhecheng
), ZHU Ju, LIU Na, REN Yanping, SUN Xiaohui, TIAN Li(), ZHANG Zhecheng
Received:2023-03-28
Revised:2023-04-10
Published:2023-07-15
Online:2023-06-20
Contact:
△TIAN Li
E-mail:yayatianli@163.com
LIU Yufei, ZHU Ju, LIU Na, REN Yanping, SUN Xiaohui, TIAN Li, ZHANG Zhecheng. Clinical characteristics of amyotrophic lateral sclerosis patients[J]. Tianjin Medical Journal, 2023, 51(7): 681-686.
CLC Number:
| 疾病进展类型 | n | 性别/ (男/女) | 起病部位/ (球部/肢体) | 起病年龄/岁 | 诊断延迟 时间/月 | 诊断级别 | ||
|---|---|---|---|---|---|---|---|---|
| 确诊 | 拟诊 | 可能 | ||||||
| 慢速进展型 | 44 | 24/20 | 8/36 | 61(55,67) | 36(18,48) | 20(45.5) | 14(31.8) | 10(22.7) |
| 中速进展型 | 37 | 19/18 | 9/28 | 60(54,64) | 18(12,24)a | 18(48.6) | 14(37.8) | 5(13.5) |
| 快速进展型 | 29 | 19/10 | 3/26 | 63(58,68)ab | 12(8,24)ab | 20(69.0) | 7(24.1) | 2(6.9) |
| χ2 | 1.425 | 2.136 | 57.781** | 19.751** | 5.938 | |||
Tab.1 Comparison of clinical parameters between ALS patients with different rates of disease progression
| 疾病进展类型 | n | 性别/ (男/女) | 起病部位/ (球部/肢体) | 起病年龄/岁 | 诊断延迟 时间/月 | 诊断级别 | ||
|---|---|---|---|---|---|---|---|---|
| 确诊 | 拟诊 | 可能 | ||||||
| 慢速进展型 | 44 | 24/20 | 8/36 | 61(55,67) | 36(18,48) | 20(45.5) | 14(31.8) | 10(22.7) |
| 中速进展型 | 37 | 19/18 | 9/28 | 60(54,64) | 18(12,24)a | 18(48.6) | 14(37.8) | 5(13.5) |
| 快速进展型 | 29 | 19/10 | 3/26 | 63(58,68)ab | 12(8,24)ab | 20(69.0) | 7(24.1) | 2(6.9) |
| χ2 | 1.425 | 2.136 | 57.781** | 19.751** | 5.938 | |||
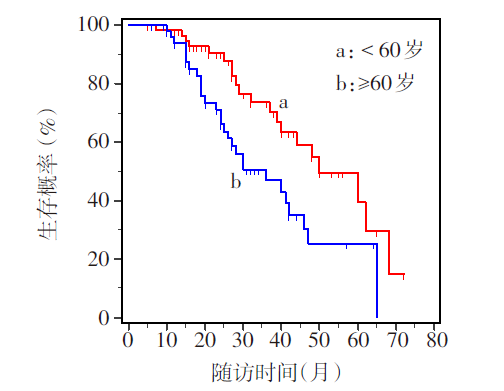
Fig.1 Survival curves of ALS patients at different ages
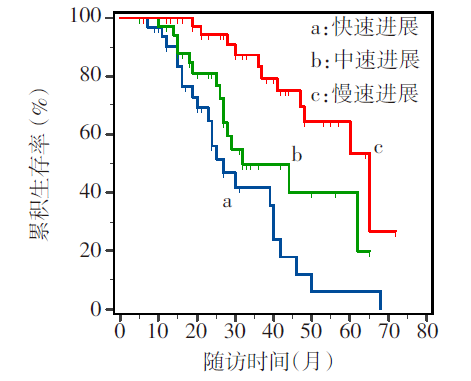
Fig.2 Survival curves of ALS patients with different rates of disease progression
| [1] | 中华医学会神经病学分会肌萎缩侧索硬化协作组. 肌萎缩侧索硬化诊断和治疗中国专家共识2022[J]. 中华神经科杂志, 2022, 55(6):581-588. |
| Amyotrophic Lateral Sclerosis Collaboration Group, Neurology Branch of Chinese Medical Association. Consensus for diagnosis and treatment of amyotrophic lateral sclerosis 2022[J]. Chinese Journal of Neurology, 2022, 55(6):581-588. doi:10.3760/cma.j.cn113694-20211212-00877. | |
| [2] | 蔡正一, 崔丽英. 肌萎缩侧索硬化症状前期研究的现状与启示[J]. 中华医学杂志, 2021, 101(7):520-523. |
| CAI Z Y, CUI L Y. Current status and implications of presymptom research on amyotrophic lateral sclerosis[J]. Natl Med J China, 2021, 101(7):520-523. doi:10.3760/cma.j.cn112137-20200529-01698.ss. | |
| [3] | BROOKS B R, MILLER R G, SWASH M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis[J]. Amyotroph Lateral Scler Other Motor Neuron Disord, 2000, 1(5):293-299. doi:10.1080/146608200300079536. |
| [4] | KIMURA F, FUJIMURA C, ISHIDA S, et al. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS[J]. Neurology, 2006, 66(2):265-267. doi:10.1212/01.wnl.0000194316.91908.8a. |
| [5] | MEHTA P, RAYMOND J, PUNJANI R, et al. Prevalence of amyotrophic lateral sclerosis (ALS), United States, 2016[J]. Amyotroph Lateral Scler Frontotemporal Degener, 2022, 23(3/4):220-225. doi:10.1080/21678421.2021.1949021. |
| [6] | CHEN L, ZHANG B, CHEN R, et al. Natural history and clinical features of sporadic amyotrophic lateral sclerosis in China[J]. J Neurol Neurosurg Psychiatry, 2015, 86(10):1075-1081. doi:10.1136/jnnp-2015-310471. |
| [7] | LOGROSCINO G, TRAYNOR B J, HARDIMAN O, et al. Incidence of amyotrophic lateral sclerosis in Europe[J]. J Neurol Neurosurg Psychiatry, 2010, 81(4):385-390. doi:10.1136/jnnp.2009.183525. |
| [8] | MANJALY Z R, SCOTT K M, ABHINAV K, et al. The sex ratio in amyotrophic lateral sclerosis: A population based study[J]. Amyotroph Lateral Scler, 2010, 11(5):439-442. doi:10.3109/17482961003610853. |
| [9] | LIU M S, CUI L Y, FAN D S. Age at onset of amyotrophic lateral sclerosis in China[J]. Acta Neurol Scand, 2014, 129(3):163-167. doi:10.1111/ane.12157. |
| [10] | LEE J W, KANG S W, CHOI W A. Clinical course of amyotrophic lateral sclerosis according to initial symptoms: An analysis of 500 cases[J]. Yonsei Med J, 2021, 62(4):338-343. doi:10.3349/ymj.2021.62.4.338. |
| [11] | PAPE J A, GROSE J H. The effects of diet and sex in amyotrophic lateral sclerosis[J]. Rev Neurol (Paris), 2020, 176(5):301-315. doi:10.1016/j.neurol.2019.09.008. |
| [12] | CHEN L, XU L, TANG L, et al. Trends in the clinical features of amyotrophic lateral sclerosis: A 14-year Chinese cohort study[J]. Eur J Neurol, 2021, 28(9):2893-2900. doi:10.1111/ene.14943. |
| [13] | XU L, LIU T, LIU L, et al. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: A systematic review and meta-analysis[J]. J Neurol, 2020, 267(4):944-953. doi:10.1007/s00415-019-09652-y. |
| [14] | CHIÒ A, CALVO A, MOGLIA C, et al. Phenotypic heterogeneity of amyotrophic lateral sclerosis: A population based study[J]. J Neurol Neurosurg Psychiatry, 2011, 82(7):740-746. doi:10.1136/jnnp.2010.235952. |
| [15] | PALESE F, SARTORI A, LOGROSCINO G, et al. Predictors of diagnostic delay in amyotrophic lateral sclerosis: A cohort study based on administrative and electronic medical records data[J]. Amyotroph Lateral Scler Frontotemporal Degener, 2019, 20(3/4):176-185. doi:10.1080/21678421.2018.1550517. |
| [16] | IWASAKI Y, IKEDA K, ICHIKAWA Y, et al. The diagnostic interval in amyotrophic lateral sclerosis[J]. Clin Neurol Neurosurg, 2002, 104(2):87-89. doi:10.1016/s0303-8467(01)00188-3. |
| [17] | RICHARDS D, MORREN J A, PIORO E P. Time to diagnosis and factors affecting diagnostic delay in amyotrophic lateral sclerosis[J]. J Neurol Sci, 2020, 417:117054. doi:10.1016/j.jns.2020.117054. |
| [18] | PAGANONI S, MACKLIN E A, LEE A, et al. Diagnostic timelines and delays in diagnosing amyotrophic lateral sclerosis (ALS)[J]. Amyotroph Lateral Scler Frontotemporal Degener, 2014, 15(5/6):453-456. doi:10.3109/21678421.2014.903974. |
| [19] | 贾蕊, 靳娇婷, 胡芳芳, 等. 221例散发型肌萎缩侧索硬化症的临床特征[J]. 西安交通大学学报(医学版), 2018, 39(5):624-628. |
| JIA R, JIN J T, HU F F, et al. Clinical characteristics of 221 cases of sporadic amyotrophic lateral sclerosis[J]. Journal of Xi'an Jiaotong University(Medical Sciences), 2018, 39(5):624-628. doi:10.7652/jdyxb201805003. | |
| [20] | CELLURA E, SPATARO R, TAIELLO A C, et al. Factors affecting the diagnostic delay in amyotrophic lateral sclerosis[J]. Clin Neurol Neurosurg, 2012, 114(6):550-554. doi:10.1016/j.clineuro.2011.11.026. |
| [21] | BORGHETTI V S, CINTRA V P, RAMOS J O, et al. Misdiagnoses in a Brazilian population with amyotrophic lateral sclerosis[J]. Arq Neuropsiquiatr, 2022, 80(7):676-680. doi:10.1055/s-0042-1755224. |
| [22] | 王惠芳, 樊东升, 张俊, 等. 肌萎缩侧索硬化症的院前误诊分析[J]. 中国现代神经疾病杂志, 2005, 5(4):240-243. |
| WANG H F, FAN D S, ZHANG J, et al. Analysis of misdiagnosis of amyotrophic lateral sclerosis pre-admission[J]. Chinese Journal of Contemporary Neurology and Neurosurgery, 2005, 5(4):240-243. doi:10.3969/j.issn.1672-6731.2005.04.010. | |
| [23] | PINTO S, SWASH M, DE CARVALHO M. Does surgery accelerate progression of amyotrophic lateral sclerosis?[J]. J Neurol Neurosurg Psychiatry, 2014, 85(6):643-646. doi:10.1136/jnnp-2013-305770. |
| [24] | KIM J M, PARK J H, KIM H S, et al. Epidemiology and diagnostic process of amyotrophic lateral sclerosis as distinct from myelopathy: 5-year cohort study of whole-population in South Korea[J]. Amyotroph Lateral Scler Frontotemporal Degener, 2018, 19(7/8):547-554. doi:10.1080/21678421.2018.1491600. |
| [25] | STATLAND J M, BAROHN R J, MCVEY A L, et al. Patterns of weakness, classification of motor neuron disease, and clinical diagnosis of sporadic amyotrophic lateral sclerosis[J]. Neurol Clin, 2015, 33(4):735-748. doi:10.1016/j.ncl.2015.07.006. |
| [26] | CHEN A, WEIMER L, BRANNAGAN T 3rd, et al. Experience with the Awaji Island modifications to the ALS diagnostic criteria[J]. Muscle Nerve, 2010, 42(5):831-832. doi:10.1002/mus.21841. |
| [27] | SHAYYA L, BABU S, PIORO E P, et al. Distal predominance of electrodiagnostic abnormalities in early-stage amyotrophic lateral sclerosis[J]. Muscle Nerve, 2018, 58(3):389-395. doi:10.1002/mus.26158. |
| [28] | FALLAT R J, JEWITT B, BASS M, et al. Spirometry in amyotrophic lateral sclerosis[J]. Arch Neurol, 1979, 36(2):74-80. doi:10.1001/archneur.1979.00500380044004. |
| [29] | EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis, ANDERSEN P M, ABRAHAMS S, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force[J]. Eur J Neurol, 2012, 19(3):360-375. doi:10.1111/j.1468-1331.2011.03501.x. |
| [30] | 樊东升, 张华纲, 邓畔. 重视和加强运动神经元病患者的早期无创正压通气治疗[J]. 中华内科杂志, 2015, 54(9):747-748. |
| FAN D S, ZHANG H G, DENG P. Pay attention to and strengthen the early non-invasive positive pressure ventilation therapy of motor neurone patients[J]. Chin J Intern Med, 2015, 54(9):747-748. doi:10.3760/cma.j.issn.0578-1426.2015.09.002. | |
| [31] | BOND L, GANGULY P, KHAMANKAR N, et al. A comprehensive examination of percutaneous endoscopic gastrostomy and its association with amyotrophic lateral sclerosis patient outcomes[J]. Brain Sci, 2019, 9(9):223. doi:10.3390/brainsci9090223. |
| [32] | 郭晓燕, 商慧芳, 方登富, 等. 中国西南地区散发性肌萎缩侧索硬化症患者的临床特征[J]. 中国神经精神疾病杂志, 2010, 36(5):296-300. |
| GUO X Y, SHANG H F, FANG D F, et al. Clinical features of sporadic patients with amyotrophic lateral sclerosis in southwest China[J]. Chin J Nerv Ment Dis, 2010, 36(5):296-300. doi:10.3969/j.issn.1002-0152.2010.05.010. | |
| [33] | WESTENENG H J, DEBRAY T, VISSER A E, et al. Prognosis for patients with amyotrophic lateral sclerosis:Development and validation of a personalised prediction model[J]. Lancet Neurol, 2018, 17(5):423-433. doi:10.1016/S1474-4422(18)30089-9. |
| [34] | DORST J, CHEN L, ROSENBOHM A, et al. Prognostic factors in ALS:A comparison between Germany and China[J]. J Neurol, 2019, 266(6):1516-1525. doi:10.1007/s00415-019-09290-4. |
| [35] | ZHANG H G, CHEN L, TANG L, et al. Clinical features of isolated bulbar palsy of amyotrophic lateral sclerosis in Chinese population[J]. Chin Med J (Engl), 2017, 130(15):1768-1772. doi:10.4103/0366-6999.211538. |
| [36] | MOGLIA C, CALVO A, GRASSANO M, et al. Early weight loss in amyotrophic lateral sclerosis: outcome relevance and clinical correlates in a population-based cohort[J]. J Neurol Neurosurg Psychiatry, 2019, 90(6):666-673. doi:10.1136/jnnp-2018-319611. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||